SimuTech Group’s engineering consultants, specializing in customized medical devices, biotechnology, and general industry expertise, are often asked by business partners, how do I get my product FDA compliant? Or better yet, is it worth it to find out?
While each product-type and use-case often has its own bits of nuance to explore, this article hopes to shed some light on basic FDA parameters, how to get approved, what products need approval (and which ones don’t), and extenuating circumstances.
In addition, we hope to make clear that this is an industry that will flourish over the coming years, and that the initial legwork required is worth your businesses time and resources in the long run. In addition, SimuTech Group’s engineering experts will alleviate your design, process, and legal hang-ups. At the end of the day, we want to see your ideas come to life, shaping the world for the better.
Federal law mandates that device manufacturers give the FDA at least 90 days’ notice of their intention to commercialize a medical device. For any new device for which there is no comparable or predicate—unless such a device may be reclassified as a “de novo” device—the FDA requires a PMA, which is the strictest device marketing application. A device must demonstrate in a PMA that there is enough scientific proof to support its safety and efficacy in the usage for which it is intended.
The FDA encourages sponsors and investigators to establish early contact and collaboration with the appropriate review division and has a pre-submission process to help PMA and 510(k) applications – these are “Q-sub” meetings, also referred to as “pre-submission” sessions.
An altered PMA may occasionally be authorized in conjunction with the FDA. One such is a “modular PMA,” which is a procedure typically used for devices at the beginning of clinical studies and entails submitting elements of the application in modules as they are finished. The “streamlined” PMA is a current FDA pilot initiative within the Division of Clinical Laboratory Devices.
If the FDA has prior experience examining devices similar to this one, this PMA may be helpful. The pre-submission procedure can help establish whether a PMA can be “streamlined” or filed as a modular application, which might significantly cut down on the time and work required to have some devices approved.
A fast-track procedure for devices is the PMN, also known as a 510(k) application, in which the sponsor demonstrates that the product is substantially equivalent to an already approved and marketed product. A predicate device in a PMN for a different, new device cannot be a device that is currently the subject of a PMA evaluation but has not yet received approval.
A PMA application is not required if the FDA finds that the device has an acceptable predicate; PMN can move forward. A complete PMA will be necessary if the FDA decides at the time the application is submitted that the proposed predicate is ineligible and rejects the 501(k) application.
For determining the substantive equivalence of a proposed predicate device, the FDA has given a decision-making algorithm. The FDA must receive PMN at least 90 days before the planned marketing date.
A humanitarian use device (HUD) is one that is meant to diagnose or cure illnesses that afflict fewer than 4K people annually in the US. HDE are managed by the FDA’s Office of Orphan Product Development.
In addition to FDA permission, the use of a HUD necessitates local IRB approval and oversight. The application for an HDE is similar to that for a PMA, with the exception that scientific proof of efficacy is not necessary due to the fact that it may take years just to recruit enough participants for a clinical research to reach statistical significance.
Demonstrating that there is an anticipated benefit to health must be shown by the device sponsor. Additionally, the product shows that the benefit is likely to outweigh the risk of disease or harm brought on by the gadget. In other words, the HDE only requires evidence of device-relative safety; device efficacy is not required.
The price the manufacturer can charge for such devices is restricted to covering manufacturing fees, research and development costs, and other clearly defined costs in order to protect particularly vulnerable patient cohorts from manufacturers who might hope to profit from devices with unproven efficacy.
A report from a qualified independent accountant attesting to the excess cost and its causes must be submitted for each equipment that costs more than $300. Devices utilized in pediatric populations are given some exceptions to this restriction.
The use of the HUD is permitted by an approved HDE, but only at institutions where a local IRB has been created to oversee device clinical testing, and only after receiving approval from the local IRB. The labeling of the item must indicate that it is a humanitarian device and that, despite being legal to use under federal law, its efficacy for the given indication has not been shown.
Understanding the approval process and prerequisites of device design requires detailed, and comprehensive knowledge, in order to best guide the client down the ‘correct path’ to market approval. This educational (and logistical) component is one of the core offerings and value proposition for seeking medical device consulting experts.
Short Answer: Perfumes, makeup, moisturizers, shampoos, hair colors, face and body cleansers, and shaving products are a few examples of consumer goods where FDA approval is not required for market entry or associated certification and labeling.
In general, compounding is a process where a pharmacist or doctor combines chemicals to make prescriptions that are tailored to the needs of specific patients, including those who have allergies to components of FDA-approved pharmaceuticals or who are unable to swallow an FDA-approved pill.
In order to assess the safety, efficacy, or quality of compounded medications, the FDA does not conduct premarket review.
The FDA’s safe and effective criteria for reviewing medical items does not apply to tobacco products since there is no such thing as a safe tobacco product. Instead, the FDA regulates tobacco products in accordance with a public health standard that takes into account the hazards the product poses to the general public, including both tobacco users and nonusers.
Manufacturers must obtain FDA approval before they may lawfully sell or distribute a new tobacco product in the United States. Premarket tobacco product applications, significant equivalence applications, and exemption from substantial equivalence requests are the three available routes for bringing a new tobacco product to market.
An approval for marketing does not imply that a tobacco product is “authorized” or safe. It indicates that the producer has fulfilled with the legal requirements to supply its goods on the open market.
Perfumes, makeup, moisturizers, shampoos, hair colors, face and body cleansers, and shaving products are a few examples of cosmetics. FDA approval is not necessary for cosmetic items, chemicals, or labeling.
One thing to note: color additives (other than coal-tar hair dyes) – Cosmetics must be appropriately labeled and safe for their intended purpose.
A medical food is designed to be taken or administered enterally, and it is meant for the specialized dietary management of a disease or condition for which precise nutritional requirements, based on accepted scientific principles, are identified by medical evaluation.
Phenylketonuria, a hereditary abnormality, is an illustration of a disease or condition that could be treated with a medical food. Medical foods that are made to be free of the amino acid phenylalanine may be necessary for someone with this disease.
A medicinal food is meant to be consumed while being closely monitored by a doctor. Products for the management of diseases as well as meal replacements and diet shakes are excluded. This would apply to conditions like diabetes (type II), which can be controlled by altering the regular diet.
FDA premarket approval is not required for medical foods. Medical food manufacturers must still abide by additional regulations, such as the registration of food facilities and current good manufacturing procedures. Medical foods are exempt from the requirement to carry a Nutrition Facts label on their packaging, but any representations made there or in other labeling must be accurate and not deceptive.
Before infant formula can be sold, the FDA does not give its approval. However, the FDA has regulatory control over infant formula producers.
The federal nutrient requirements and other laws must be complied with by manufacturers of baby formula. Before releasing a novel formula, manufacturers must submit an infant formula submission to the FDA and register with the organization.
Every year, the FDA inspects all factories that produce infant formula, as well as collecting and examining product samples. New facilities are also inspected by the FDA. The manufacturer of the formula is required to initiate a recall if the FDA determines that adulterated or misbranded infant formula poses a danger to human health.
Health care providers, including doctor’s offices and laboratories, are not “approved” by the FDA. The FDA is allowed to check regulated facilities to make sure they adhere to the most recent good manufacturing practices.
Although the agency frequently inspects production facilities and contract manufacturers as part of a product application for some items that need premarket approval, the agency does not independently approve manufacturing facilities.
The FDA is not permitted to certify the efficacy and safety of dietary supplements. In fact, the FDA need not even be informed when marketing numerous dietary supplements.
However, businesses must notify the FDA of premarket safety concerns at least 75 days. That is, prior to marketing dietary supplements made with certain “novel dietary components” (that were not marketed in the U.S. before Oct. 15, 1994).
Before putting their products on the market, dietary supplement manufacturers must make sure they are safe. In addition, that they also adhere to other labeling and quality standards, like good manufacturing practices.
The Nutrition Facts label and other food labels are not approved by the FDA.
Contrary to popular belief, the FDA does not authorize specific food labels. In effect, FDA adherence has no bearing on how consumer goods can be marketed. However, according to FDA regulations, this may differ for particular labeling components. For example, nutritional data must be present on dietary supplements. Any comments made about food goods must also be “accurate” and not deceptive. In addition, maintain adhere to any regulations that may be applicable for the particular statement.
The “Nutrition Facts” label (or the “Supplement Facts” label for dietary supplements) is required to list two items. That is, the serving size of the food and specific details on the nutrients contained in each serving. In addition, the “Use By” date that your dad checks is note approved or determined by the FDA. These are determined (sometimes arbitrarily) by the manufacturer.
A medical device takes an average of 3 to 7 years to reach the market, whereas pharmaceutical medications take an average of 12 years.
There are worries that the Food and Drug Administration’s procedures would not be adequate to provide the necessary assurances of safety and efficacy, according to Dr. Gail A.Van Norman’s article, Drugs, Devices, and the FDA, published at Department of Anesthesiology and Pain Medicine, University of Washington, Seattle, Washington, USA.
Given SimuTech Group extensive knowledge of the medical and device regulatory environment, we can help to significantly reduce this time by ensuring the proper arrangements and protocols are developed at the beginning of the design stage.

We pride ourselves in being responsive to each client’s particular needs, from initial, quick concept development analyses to in-depth, highly complex multi-disciplinary analyses, all within FDA approval.
Medical Device Consulting Engineers
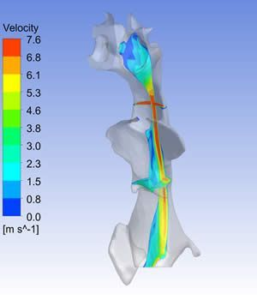
Centrifugal Blood Pump Modeling
For over ten years, the SimuTech Group has applied CFD in healthcare simulation for the design of left and right ventricular assist devices as well as total artificial hearts. Our focus in medical device consulting has been to provide insights guiding the development of these new blood pumps. The end-goal being to provide the desired hydraulic performance and also minimize regions of sustained high and low shear that could lead to blood damage or thrombus formation.
Patient-Specific Computational Modeling
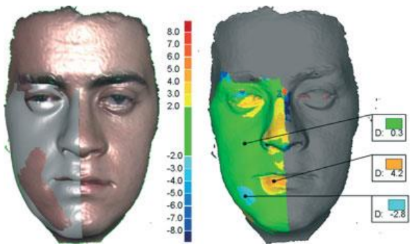
An exciting and growing area of focus in healthcare is patient-specific computational modeling. Such modeling, based upon three-dimensional imaging reconstruction of a patient’s anatomy, can help clinicians plan their surgical approaches or evaluate a new device’s performance under simulated clinical conditions.
Click here to learn more about patient-specific computational modeling.
Artificial Kidney and Pancreas Projects
Another area of focus for SimuTech Group relating to medical device consulting, has been the application of CFD to help advance the development of new artificial kidney and pancreas devices. In addition to optimizing the flow path to improve mass transfer, we have applied particle tracking methods to assess the likelihood of platelet activation when flowing through such medical devices.
Read how SimuTech Group is supporting the artificial kidney project.
Pumps & Oxygenators
Catheters & Devices